

Getting Started and Completing the Forms
What qualifies as Human Subjects Research requiring IRB Review?
For an activity to be considered human subjects research requiring the University of Texas at Austin IRB’s oversight, the activity must:
- be research
- involve human subjects or information about living individuals, and
- engage the institution in the activity (e.g. through agents/employees/affiliates conducting research, agreements, funding etc.)

What is considered research?
Department of Health and Human Services (DHHS)
Research, as defined by DHHS means a systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge (45 CFR 46.102(d)).
- Systematic Investigation is a planned activity involving qualitative or quantitative data collection and data analysis that sets forth an objective(s) and a set of procedures intended to reach the objective(s), i.e., to acquire knowledge, develop a theory, or answer a question.
- Generalizable Knowledge means information from which one may infer a general conclusion; knowledge brought into general use or that can be applied to a wider or different range of circumstances. For example, publication and presentation are typical methods used to disseminate research findings, thereby contributing to “generalizable knowledge.” However, not all information that is published or presented represents generalizable knowledge. Generalizable knowledge is also interpreted to include data intended for general use, regardless of its eventual distribution or acceptance.
Food and Drug Administration (FDA)
Research as defined by the FDA means any experiment that involves a test article and one or more human participants, and that either must meet the requirements for prior submission to the FDA under section 505 (i) or 520 (g) of the Federal Food, Drug, and Cosmetic Act, or need not meet the requirements for prior submission to the FDA under these sections of the Federal Food, Drug and Cosmetic Act, but the results of which are intended to be later submitted to, or held for inspection by, the FDA as part of an application for a research or marketing permit.
The terms research, clinical research, clinical study, study, and clinical investigation are synonymous for purposes of FDA regulations (21 CFR 50.3(c), 21 CFR 56.102(c)).
What research involves Human Subjects or information about living individuals?
Human Subjects has reference to two definitions defined by federal agencies:
- DHHS defines human subject as a living individual about whom the investigator conducting research:
- Obtains information or biospecimens through intervention or interaction with the individual and uses, studies, or analyzes the information or biospecimens; or
- Obtains, uses, studies, analyzes, or generates identifiable private information or identifiable biospecimens.
- FDA defines human subject as an individual who is or becomes a participant in research, either as a recipient of the test article or as a control. A subject may be either a healthy individual or a patient. When medical device research involves in vitro diagnostics and unidentified tissue specimens, the FDA defines the unidentified tissue specimens as human subjects.
According to HHS regulations, the University becomes engaged in human subjects research when its employees or agents (i) intervene or interact with living individuals for research purposes, or (ii) obtain individually identifiable private information about those individuals for research purposes.
- Under FDA regulations, the institution becomes engaged in human subjects research when it undertakes a clinical investigation on individuals who are or become subjects in the investigation, either as recipients of a test article or as controls and may be either patients or healthy non-patients.
- Principle Investigators are automatically considered to be “engaged” in human subjects research whenever they apply for or receive a direct award to support research that includes human subjects, even if all the activities involving human subjects will be carried out by a subcontractor or collaborator. In all cases the institution to which the grant has been awarded bears the responsibility for protecting human subjects under the award.
If you have any questions about the above and how it applies to your proposed research activity prior to submission, please contact the UT Austin IRB at IRB@austin.utexas.edu. If your research activity has already been submitted, please reach out to your assigned IRB Analyst directly via comment within UTRMS, or by email.
References/Resources:
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 2: Definitions.
- The Code of Federal Regulations - 21 CFR 50.3(c), 21 CFR 56.102(c), 45 CFR 46.102(d)
Are quality improvement efforts or program evaluations considered human subjects research?
Yes and no. There are types of Quality Improvement (QI)/Program Evaluations that may qualify as human subjects research and would require UT Austin IRB Review, and types that would not be considered human subjects research.
To be considered human subjects research the activity still needs to meet the definition of research and involve human subjects (See FAQ: What qualifies as Human Subjects Research requiring IRB Review?).
Intent
- Research: Develop or contribute to generalizable knowledge (e.g., testing hypotheses)
- Quality Improvement: Improve a practice or process within a particular institution or ensure it confirms with expected norms
- Program Evaluation: Improve a specific program
Design
- Research: Systematic; follows a rigid protocol that remains unchanged throughout the research; may involve randomization; May involve significant deviation from standard practice
- Quality Improvement: Adaptive, iterative design; may or may not be systematic; generally does not involve randomization; Unlikely to involve significant deviation from standard practice
- Program Evaluation: Adaptive, iterative design; may or may not be systematic; generally individuals not randomized; may involve comparison of variations
Mandate
- Research: Activities not mandated by institution or program (e.g., activity would not occur separately from research activity)
- Quality Improvement: Activity mandated by the institution or clinic as part of its operations (e.g., activity would still occur regardless of proposed research component)
- Program Evaluation: Activity mandated by the program, usually its funder, as part of its operations (e.g., activity would still occur regardless of proposed research component)
Effect on Program/Practice under Study
- Research: Findings are not expected to directly affect institutional or programmatic practice
- Quality Improvement: Findings are expected to directly affect institutional practice and identify corrective action(s) needed
- Program Evaluation: Findings are expected to directly affect the conduct of the program and identify improvements
Population
- Research: Usually involves a subset of individuals; no obligation to participate; may involve statistical justification of sample size to achieve endpoints
- Quality Improvement: Responsibility to participate as a component of the practice or process; information on all or most involved in the practice or process is expected to be included; exclusion of information from some individuals significantly affect conclusions
- Program Evaluation: Responsibility to participate as a component of the program; information on all or most involved in the program or treatment; exclusion of information from some individuals significantly affect conclusions
Benefits
- Research: Participants may or may not benefit directly; often a delayed benefit to future knowledge or individuals
- Quality Improvement: Directly benefits a practice, process, or system; may or may not benefit participants
- Program Evaluation: Evaluation concentrates on program improvements or whether the program continues; no expected benefit participants
Dissemination of Results*
- Research: Intent to disseminate results generally presumed at the outset of project as part of professional expectations, obligations; results expected to develop or contribute to generalizable knowledge by filling a gap in scientific knowledge or supporting, refining, or refuting results from other research studies
- Quality Improvement: Intent to disseminate results generally not presumed at the outset of the project; often does not occur beyond the institution evaluated; when published or presented to a wider audience, the intent is to suggest potentially effective models, strategies, assessment tools or provide benchmarks rather than to develop or contribute to generalizable knowledge
- Program Evaluation: Intent to disseminate results generally presumed at the outset of the project; dissemination of information to program stakeholders and participants; may be publicly posted (e.g., website) to ensure transparency of results; when published or presented to a wider audience, the intent is to suggest potentially effective models, strategies, assessment tools or provide benchmarks rather than to develop or contribute to generalizable knowledge
*Please Note: The intent to publish is an insufficient criterion for determining whether a quality improvement activity/Program Evaluation involves research. Some journals will require an official IRB determination letter regardless of the activity being human subjects research. In this case, an application will need to be submitted to the IRB in order to receive a “not human research” determination.
Examples of QI activities that are likely NOT research include:
- Implementing a practice to improve the quality of patient care
- Collecting patient or provider data regarding the implementation of the practice for clinical, practical, or administrative purposes
- Measuring and reporting provider performance data for clinical, practical, or administrative uses
- A group of affiliated hospitals implements an application to reduce prescription amount errors, and collects patient prescription information from medical charts to assess whether the application helped reduce error rates as expected.
Examples of Activities that are likely QI and Research:
- A project involves introducing an untested clinical intervention for purposes which include not only improving the quality of care but also collecting information about patient outcomes for the purpose of establishing scientific evidence to determine how well the intervention achieves its intended results.
- Collaborative (multi-site) – All the sites are trying to improve some aspect of clinical care (ex. implementing an application (app) to help improve making clinical decisions). The whole department decides this app will improve care, and implement the app. They collect data as the app is implemented, and in addition, analyze this data for generalizable knowledge.
- A teacher implements a practice to have all students reflect on their learning by keeping a journal, with the intention of improving teaching practice. However, the teacher also wants to prove that this method works, so they analyze student journals with grades to generalize the success of this method.
Additional resources to assist in determining if your activity is human subjects research requiring review:
U.S. Department of Health and Human Services, Office for Human Research Protections, Quality Improvement Activities FAQs: https://www.hhs.gov/ohrp/regulations-and-policy/guidance/faq/quality-improvement-activities/index.html
UT Austin IRB, Quality Improvement Project Assessment Form: https://redcap.prc.utexas.edu/redcap/surveys/?s=WL8MXEYXRT
References:
The above is adapted in part from Virginia Commonwealth University IRB’s guidance, Quality Improvement vs. Research – Do I Need IRB Approval?, and The University of Wisconsin-Madison Health Sciences IRB’s Comparison of the Characteristics of Research, Quality Improvement, and Program Evaluation Activities.
Do students' academic research/class projects projects require IRB approval?
All activities meeting any of the definitions of human subjects research conducted under the auspices of UT Austin must be reviewed by the Institutional Review Board (IRB) prior to the start of the activity. The Office of Research Support and Compliance (RSC) recognizes that some student classroom activities involve questionnaires, interviews, or other interactions with individuals that, in a different context, might meet the definition of human subjects research. It is RSC policy that classroom activities where the purpose is to teach students research techniques or methodology with no intention to develop or contribute to generalizable knowledge do not require IRB review. More information can be found within the Student Class Projects Guidance document.
Of note, the University requires all research to be conducted under the guidance of a qualified Principal Investigator (PI). As a teaching institution, students will be engaged in conducting research as an integral part of their educational experience. Faculty who assume PI responsibility for student-led research involving human subjects must be willing to provide oversight to the research activities and assume full responsibility for the conduct of the research. If you are unsure if you are eligible to serve as a Principal Investigator, please review the eligibility table here.
If you have any questions about the above and how it applies to your proposed research activity prior to submission, please contact the UT Austin IRB at IRB@austin.utexas.edu. If your research activity has already been submitted, please reach out to your assigned IRB Analyst directly via comment within UTRMS, or by email.
References/Resources:
UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 3.12.2 Class Projects and Section 3.16.2 Faculty Principal Investigators for Student-Led Research.
Do oral history projects, case studies, or journalism require IRB Review?
To determine whether oral history or other activities solely consisting of open-ended qualitative type interviews are subject to the University’s human research protections policies, the activity must meet the following standards and general principles for evaluating oral history type activities:
- The activity involves a prospective research plan which incorporates data collection, including qualitative data, and data analysis to answer a research question; and
- The activity is designed to draw general conclusions (i.e., knowledge gained from a study may be applied to populations outside of the specific study population), inform policy, or generalize findings.
- Systematic investigations involving open-ended interviews that are designed to develop or contribute to generalizable knowledge (e.g., designed to draw conclusions, inform policy, or generalize findings) would constitute “research” as defined by 45 CFR 46. For example, an open-ended interview of surviving Gulf War veterans to document their experiences and to draw conclusions about their experiences, inform policy, or generalize findings would require IRB review and approval.
- Oral historians and qualitative investigators may want to create archives for the purpose of providing a resource for others to do research. Since the intent of the archive is to create a repository of information for other investigators to conduct research as define by 45 CFR 46, the creation of such an archive would constitute research under 45 CFR 46. For example, open ended interviews are conducted with surviving Negro League Baseball players in order to create an archive for future research. The creation of such an archive would constitute research under 45 CFR 46 since the intent is to collect data for future research.
Conversely, oral history activities, such as open-ended interviews, that only document a specific historical event or the experiences of individuals without intent to draw conclusions or generalize findings would not constitute “research” as defined by 45 CFR 46. For example, an oral history video recording of interviews with Holocaust survivors is created for viewing in the Holocaust Museum. The creation of the video does not intend to draw conclusions, inform policy, or generalize findings. The sole purpose is to create a historical record of specific personal events and experiences related to the Holocaust and provide a venue for Holocaust survivors to tell their story.
Please see for more information. Researchers are advised to consult with the IRB staff regarding whether their oral history project requires IRB review and approval at IRB@austin.utexas.edu.
References/Resources:
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 3.12.3 Oral History Projects
- HRP-UT981-Guidance Student Class Projects
- The Code of Federal Regulations - 45 CFR 46
I am planning to conduct a pilot study; do I need to submit my proposal?
Pilot studies that meet the definition of research involving human subjects require IRB review. However, if the study does not meet the definition of research involving human subjects it would not require IRB review.
Definitions can be found in the response to the FAQ: What qualifies as Human Subjects Research requiring IRB Review?
If you have any questions about the above and how it applies to your proposed project, please contact the UT Austin IRB at IRB@austin.utexas.edu. If your research activity has already been submitted, please reach out to your assigned IRB Analyst directly via comment within UTRMS, or by email.
How do I submit documents for review?
A complete application must be submitted to the UT Austin IRB through the electronic submission system, UT Research Management Suite – IRB Module (UTRMS-IRB). The Getting Started and Creating a New Study Submission (PDF) document provides step by step instructions on how to create and attach submission documents. Study proposal templates have been created by the UT Austin IRB and are available to download from the UTRMS-IRB Library, Templates tab (Figure 2).
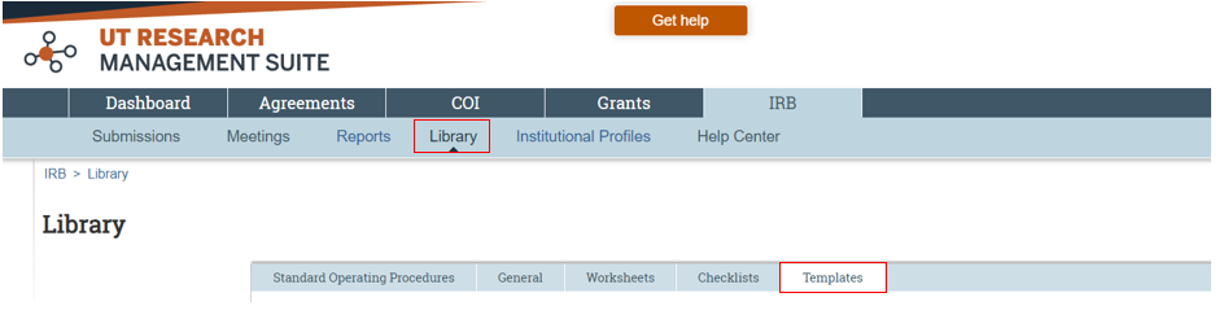
What documents should be included in the online IRB application submitted via UTRMS-IRB?
The following materials should be submitted with an application for greater than minimal risk research or otherwise requiring review by the IRB Committee at a convened board meeting, as applicable:
- A research proposal using HRP-UT901 – Template IRB Proposal Standard Submission or HRP-UT903 Template IRB Proposal Secondary Use Submission form.*
- Informed consent documents, parental permission forms, minor assent forms, as appropriate.
- Recruitment materials; i.e., flyers, posters, web-pages, email messages, etc.
- Copies of all measures/instruments if the study involves the use of questionnaires, surveys, or similar instruments.
- Data and Safety Monitoring Plan/Board (DSM-P/B), as applicable
- Site letters for extramural research.
- Review/confirmation of Environmental Health and Safety (EHS), Institutional Animal Care and Use Committee (IACUC), and/or Institutional Biosafety Committee (IBC) approval.
- Sponsor protocol (and the Investigator’s Brochure, when one exists).
- A multicenter research protocol.
- DHHS-approved protocol and/or sample consent document (when one exists).
- Supplemental forms as applicable.**
The following materials should be submitted with an application for an expedited review, as applicable:
- A research proposal using HRP-UT901 – Template IRB Proposal Standard Submission or HRP-UT903 Template IRB Proposal Secondary Use Submission form.*
- Informed consent documents, parental permission forms, minor assent forms, as appropriate.
- Recruitment materials; i.e., flyers, posters, web-pages, email messages, etc.
- Copies of all measures/instruments if the study involves the use of questionnaires, surveys, or similar instruments.
- Site letters for extramural research.
- EHS, IACUC, IBC approval documentation.
- Sponsor protocol.
- Supplemental forms as applicable.**
The following materials should be submitted with an application for an exempt determination, as applicable:
- A research proposal using HRP-UT902 – Template IRB Proposal Exempt Submission or HRP-UT903 – Template IRB Proposal Secondary Use Submission form.*
- Unless the study involves only the use of secondary data, an informed consent document/information sheet using this template or create your own that containers the following:
- disclosure to participants that the activity involves research,
- a description of the research procedures,
- who to contact with questions, and
- notice that participation is voluntary.
- Recruitment materials; i.e., flyers, posters, web-pages, email messages, etc.
- Copies of all measures/instruments if the study involves the use of questionnaires, surveys, or similar instruments.
- Site letters for extramural research.
- Supplemental forms as applicable.**
The following materials should be submitted with an application for a Not Human Subjects Research determination (if required for publication or funding source), as applicable:
- A research proposal using HRP-UT902 – Template IRB Proposal Exempt Submission form or HRP-UT903 – Template IRB Proposal Secondary Use Submission form.*
*When to use HRP-UT903 – Template IRB Proposal Secondary Use Submission form: Use only when the research involves secondary use of data that was originally created for purposes unrelated to the proposed research activity.
**When Supplemental Forms Should be Included in Application/Submission: Available supplemental forms are applicable to research involving biospecimens, investigational devices, investigational drugs/biologics, protected health information, international research, prisoners, registry or repository data, and U.S. Department of Defense are available for download from the UTRMS Library.
References/Resources:
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 3: General Policies and Procedures.
I believe my study meets the criteria for Expedited Review. Does this mean my proposal will be reviewed and approved quicker?
No, an activity receiving Expedited Review does not mean that the review will occur faster or slower than other submissions. Expedited Review applies to research activities that have been determined to be minimal risk and fits one or more of the categories authorized by 45 CFR 46.110 for expedited review. Please see Section 5.3 Research Appropriate for Expedited Review of HRP-UT1000 IRB Policies and Procedures Manual for more information.
References/Resources:
- The Code of Federal Regulations - 45 CFR 46.110.
Who should be listed as Local Study Team Members?
All faculty, staff, and students who are or will be involved in the conduct of human subjects research should be listed as Local Study Team Members within the UTRMS study record upon initial submission or through modifications (for non-exempt research).
This applies to all persons with a significant role in the research (e.g., Principal Investigators, Co-Investigators, Project Managers, or Research Assistants) and designated to:
- Recruit potential research participants,
- Obtain informed consent from prospective research participants,
- Interact or intervene with participants to collect research data, or
- Analyze identifiable research data.
All researchers affiliated with UT Austin should automatically be available to select from the personnel pick list. If you have any questions, and the activity is already under review with an assigned analyst, please post a comment to the UTRMS-IRB study workspace with your questions and select “IRB Coordinator” to be notified of its posting.
Human Subjects Research CITI Training
University policy requires training for all faculty, faculty mentors, researchers, and students, including researchers from other institutions who wish to conduct human subjects research at the University. All personnel, originally listed or later added to a study through an amendment/modification, must complete human subjects research training. In order to comply with the policy, researchers are required to complete the University’s training affiliated with Collaborative Institutional Training Initiative (CITI). Completion of this training must be accomplished every three years.
Financial Conflict of Interest Disclosures
Principal Investigators (PIs) and their research staff are required to disclose financial interests in UTRMS-COI according to the University Conflicts of Interest Policy found in the Handbook of Operating Procedures and Policy Memoranda (7-1210, 7-1220).
- Exempt research: PIs who have a potential financial conflict of interest* relating to the submitted study are required to submit Research Certifications in UTRMS-COI confirming financial disclosures are current.
- Non-exempt research (i.e., expedited and full board research): PIs and research staff identified by the PI as Covered Individuals* are required to submit Research Certifications in UTRMS-COI confirming financial disclosures are current.
*Covered Individuals are defined as individuals who, regardless of title or position, have decision making authority related to the design, conduct, reporting, review, or oversight of research (HOP 7-1210).
References/Resources:
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual; Section 3.8.4 Principal Investigator and Research Staff, Section 3.15 Training Requirements.
- UT Austin’s Handbook of Operating Procedures and Policy Memoranda (HOP) 7-1210 and 7-1220.
- IRB Training webpage.
- Conflict of Interest Program webpage.
- Who is a Covered Individual? document.
What is the best way to view determinations if a study or modification/continuing review was reviewed at a convened board meetin
Disclaimer: This question and response applies only to research that is greater than minimal risk or otherwise requires review by the Committee at a convened Institutional Review Board Meeting.
Once a research activity has been reviewed and a determination made by the IRB Committee, correspondence will be sent via email notifying the Principal Investigator as well as the listed Primary Contact or PI Proxy (if applicable) of the determination.
One of the following determinations will be made:
- Approved: Made when all criteria for approval are met. Approved by the IRB as written with no explicit conditions.
- Approved with Explicit Conditions or Modifications Required to Secure Approval: Approved with requirements for minor changes or simple concurrence of the PI. These will be identified to the PI and must be completed and documented prior to beginning the research. Minor or prescriptive changes or requirements will be reviewed for approval by the IRB chair or a designated IRB member, typically the IRB staff person assigned to the study. Substantive changes or requirements, requests for more information for IRB consideration, and other issues related to the criteria for approval require review and approval by the convened IRB.
- Deferred or Tabled: Generally, the protocol or consent form has deficiencies that prevent accurate determination of risks and benefits or requires significant clarifications, modifications, or conditions that, when met or addressed, require full IRB review and approval of the PI’s responses and revisions. The deficiencies will be specified to the PI, and on occasion the PI is asked to attend the full board meeting in order to clarify the points in question. The PI must revise the protocol, consent forms, or other documents as specified by the IRB and re-submit the entire protocol for full review at a convened meeting. The PI may request reconsideration of determination by submitting a written response to the IRB. The IRB will invite the PI to the IRB meeting if the IRB has additional questions. The IRB will reconsider its original decision in light of new information presented by the PI.
- Disapproved: This determination may only be made at a convened IRB meeting. The protocol describes a research activity that is deemed to have risks which outweigh potential benefits or the protocol is significantly deficient in several major areas. The protocol and/or other documents will need to be completely re-written and re-submitted as a new submission. PIs may request reconsideration of disapproved studies by submitting a written response to the IRB. The IRB will invite the PI to the IRB meeting if the IRB has additional questions. The IRB will reconsider its original decision in light of new information presented by the PI. The second decision is final.
Activities that are approved without required modifications to secure approval will populate the study record with initial approval, effective and approval end dates within the top right corner of the main/parent study page in UTRMS (Figure 3).
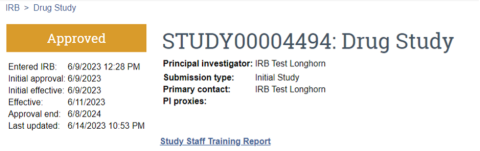
If determinations other than approval have been made, the top left corner of the main/parent study page in UTRMS will reflect the current status (Figure 4).

If you have any questions, and the activity is already under review with an assigned analyst, please post a comment with your questions and select “IRB Coordinator” to be notified of its posting. For general inquiries, please contact the UT Austin IRB at IRB@austin.utexas.edu.
References/Resources:
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 5.5 Possible IRB Protocol Determinations.
When should I submit my proposal?
Please budget at least two to three weeks for Exempt Review and three to four weeks for Expedited Review. Greater than minimal risk research or activities otherwise requiring review by the IRB Committee at a convened board meeting must be submitted by the agenda deadline date to be added for review at a specific meeting date (approximately one month in advance). Please review the published agenda and meeting dates for the convened board.
What is required for off-campus/extramural research?
Research that is physically conducted on private property (schools, hospitals, community agencies, NGOs, businesses) requires documented approval from that site (site letter). A site approval letter should:
- Be written on official letterhead by the person most responsible for activities being conducted at the site location.
- Provide attestation that the person most responsible is informed about the research and approves the conduct of the research at the site location.
For research occurring at or using Ascension Seton facilities/resources/data, confirmation of a Seton Site Approval Tool (SAT) submission is required as part of your IRB Submission/Application. Specifically, we require the confirmation email that states the request has been submitted as Seton will require the IRB determination letter prior to their own approval. This applies to all Ascension Seton clinics, Dell Children’s Medical Center, Dell Seton Medical Center at UT Austin and you can complete the SAT submission here.
When performing human subjects research physically in other countries, University researchers are expected to comply with U.S. regulations and guidelines and any applicable regulations of the country in which the research is performed. Researchers demonstrate whether the university or researcher has permission to conduct research in the country by local ethics committee review and approval or by certification (approval) by the local government when there is no local ethics committee. Researchers conducting transnational research are required to complete and upload a copy of HRP-UT908 Template IRB Supplemental Form International Research, located within the UTRMS-IRB Library. For assistance in determining applicable international research protections, please access the U.S. Department of Health and Human Services, Office for Human Research Protections, International Compilation of Human Research Standards.
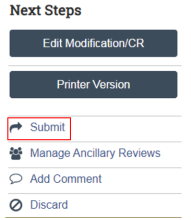
My study is still in Pre-Submission and I don’t see a Submit option. What should I do?
This is a common issue encountered for student-led research projects that require oversight of the research by an eligible Principal Investigator (PI). If you do not see a submit option it is likely that you are not the listed PI of record or assigned PI Proxy status (a listed study team member who has been assigned by the PI to act on behalf of the PI). Only the PI and the PI Proxy have access to the “Submit” function on the left side of the study page underneath “Next Steps” (Figure 6). If this situation applies to you, please reach out to the listed PI to either submit the application or assign you as PI Proxy following the instructions here.
Are there any costs for UT IRB review?
If the research activity is an industry sponsored clinical trial or multisite research where UT Austin IRB is the reviewing IRB or is deferring oversight, then there may fees involved.
References/Resources:
My study might involve the use of deception; what do I need to consider?
Research involving deception and incomplete disclosure involves intentionally communicating information to research subjects in a way that produces false beliefs. Obfuscation or withholding information at the outset of a study is also considered deception. Any research in which information is withheld until subjects have participated to some degree should be considered as a deception study.
The following are general guidelines regarding the design, review and conduct of studies involving deception and incomplete disclosure:
- Use of deception and incomplete disclosure is usually only acceptable for studies that are minimal risk.
- The use of deception/incomplete disclosure should have no adverse effects on the well-being of subjects.
- The IRB must be supplied with sufficient information to determine that the value of the research outweighs the risk of waiving some aspects of the requirement for full disclosure in the informed consent process. (See Section 6.7 Waiver of Informed Consent and Waiver of Documentation of Consent)
- There is no reasonable alternative to scientifically and effectively address the research question without the use of deception/incomplete disclosure.
- Subjects are not deceived about any aspect of the study that would alter their willingness to participate.
- As soon as it is appropriate, debriefing should be accomplished and the deception/incomplete disclosure explained to subjects.
- When appropriate, subjects should be informed prospectively of the use of deception/incomplete disclosure and consent to its use.
- During debriefing inform subjects of their right to withdraw their data, if they wish, and how that will be accomplished.
To assist the IRB in its review and determination of the appropriateness of the research study, Principal Investigator(s) should address the following items in the relevant documents:
- Explain the reason(s) for use of deception/incomplete disclosure in the study design. Specifically, address why complete disclosure would compromise the scientific validity of the study.
- Describe the extent of the deception/incomplete disclosure in detail and how it relates to the study aims and design.
- Justify and discuss how the proposed research, involving deception/incomplete disclosure involves no more than minimal risk to subjects. Consider all levels of increased risk subjects could experience as a result of the deception/incomplete disclosure methodology.
- Justify and discuss why there are no feasible or scientifically valid alternative methods, which do not involve deception/incomplete disclosure, to conduct the research.
- Describe the methods for prompt disclosure to debrief subjects. This should be accomplished as soon as possible after subjects complete research related activities. Also describe how you will assure that subjects leave the study setting with a clear and accurate understanding of the deception/incomplete disclosure and the reasons for using this methodology. If debriefing is not planned, justify why.
References/Resources:
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 15: Research Using Deceptive or Incomplete Disclosure.
I will be collaborating with another institution. Do I need to submit to UT’s IRB and the other institution?
Typically, UT Austin researchers must receive UT Austin IRB approval to conduct research with human subjects, regardless of where the research takes place. When collaborative projects are expected to involve researchers from multiple institutions, contact the IRB Reliance Office to determine next steps (IRBreliance@austin.utexas.edu). While, in some cases, each investigator should work with their own institution’s IRB, in other cases it might be desirable (or required by sponsor or funding agency) to arrange a Reliance agreement to designate one IRB to review and approve the research as a whole.
References/Resources:
- UT Austin IRB’s Reliance Guidance
My research will be done in another country; Do I have to obtain IRB review and approval from UT Austin prior to conducting rese
Yes, UT Austin researchers must receive UT Austin IRB approval or exempt determination to conduct research with human subjects, regardless of where the research takes place. You should also be aware that your project may need local IRB approval (or the equivalent ethical review), in addition to UT Austin IRB’s determination. If the research will be conducted outside the United States, please be sure to fill out the HRP-UT908 - Template IRB Supplemental Form International Research form, located within the UTRMS-IRB Library.
Disclaimer: If you are or will be conducting research outside of the United States, please be aware that UT currently has an international travel policy that involves approval for student/faculty/staff travel to areas of high risk. Please refer to the official UT Travel Policy to Restricted Regions for more details, and to see whether your travel requires review.
I am planning on holding a drawing for my compensation method; what should I consider?
The key question for researchers who want to use drawings as incentives for research participation is whether the subject’s participation requires a substantial amount of time and effort. Researchers should ensure that no conditions are imposed for enrollment. This means that everyone is eligible for the drawing upon providing consent to participate in the study. If the researcher wants to impose a condition (e.g., completion of the survey) before entry is granted to the subject, then the IRB (with legal counsel if needed) will need to make the determination as to whether the condition(s) involve a “substantial expenditure of time and effort” on behalf of the subject.
The determination of “substantial time and effort” is made on a case-by-case basis and considers various factors such as the time involvement of the subject, if return visits are required, what is asked of the subject (e.g., survey completion, blood draw), etc.
References/Resources:
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 4.8.2 Use of Lotteries.
Consent & Recruitment
What is Informed Consent?
Researchers should consider obtaining informed consent as a process, not just a consent form, by which the research study is thoroughly explained to the potential subject. The requirement to obtain informed consent should be seen as not only a legal obligation, but also as an ethical obligation. Documentation of informed consent is accomplished through the use of a consent form. Prior to enrolling subjects in a research activity, researchers are required to obtain legally effective informed consent from a potential subject or their Legally Authorized Representative (LAR) and, if the research involves children, a parent’s permission or child’s consent.
Exceptions to Informed Consent Requirements:
Exempt Research:
- Federal regulations for informed consent do not apply to exempt research, however, there is an institutional requirement for an informed consent process for exempt research. For exempt research where there are interactions with subjects, the following is required:
- There will be a consent process.
- The consent process will disclose that the activities involve research.
- The consent process will disclose the procedures to be performed.
- The consent process will disclose that participation is voluntary.
- The consent process will disclose the name and contact information for the investigator.
- There are adequate provisions to maintain the privacy interests of subjects.
Note: Documentation of consent is not a required element and signatures are not required to be collected. Researchers may create their own consent document/information implementing the above requirements or the HRP-UT926 - Template Exempt Information Sheet available within the UTRMS Library.
Expedited or Greater than Minimal Risk Research:
- Informed Consent is required unless a waiver or alteration of some or all of the elements is requested by the researcher and the waiver is approved by the IRB.
- Written documentation of informed consent (written signature of the subject) is required unless the research meets the criteria for waiver of documentation.
Note: Waiver/Alteration applications are included within sections the HRP-UT901 – Template IRB Proposal Standard Submission form.
References/Resources:
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 6: Informed Consent.
- See UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 6.7 Waiver of Informed Consent and Waiver of Documentation of Consent for more information about waivers and alterations.
- HRP-UT901 – Template IRB Proposal Standard Submission, HRP-UT920a - Template Informed Consent Form English, HRP-UT921a - Template Parental Permission Form English, HRP-UT922a - Template Assent Form English, HRP-UT926 - Template Exempt Information Sheet from the UTRMS Library.
Who can provide consent on behalf of a subject for participation in a research study?
The regulations are clear that written documentation of informed consent (or permission of the parents if the subject is a child) of the subject (or Legally Authorized Representative) is required for non-exempt research unless a waiver or alteration is approved by the IRB.
If potential subjects are deemed as decisionally impaired, informed consent must be obtained from a Legally Authorized Representative (LAR). They should be told that their obligation is to try to determine what the subject would do if they were competent, or if the subject’s wishes cannot be determined, what they think is in the best interest of the decisionally impaired subject. The IRB must approve the inclusion of decisionally impaired subjects. When a PI proposes to conduct a research project utilizing adult subjects who by virtue of age, physical impairment, mental impairment, or any other reason may not be able to personally execute legally effective informed consent, the IRB shall review the project on the basis of risk and benefit.
References/Resources:
- The Code of Federal Regulations, 45 CFR 46.
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 6: Informed Consent, Section 12.4 Research Involving Children.
Can I recruit students for my research from my own class?
We prefer that you do not use students from your own class as subjects because of the inherent power relationship (e.g., through their responsibility for assigning grades) where it is likely that some students will feel pressure to comply with requests made by their instructors. In other words, the primary issue with gathering data from one’s own course is the potential for undue influence.
In the rare instances in which recruiting from one’s own class is permissible, researchers are expected to minimize the potential for students to feel pressured to participate. There are various strategies for minimizing the potential pressure to participate. One way that researchers have reduced the potential to cause undue influence is to design the study so that the instructor is blind to the identity of the participants (at least until after the final grades have been assigned). For example, a researcher can run the study and keep any identifying information from the instructor. If a researcher designs a study in this way these points are crucial:
Before being asked to participate, potential subjects should be informed that the instructor will not know who did and who did not participate (at least until after the final grades have been assigned).
The research should be designed so that the instructor cannot infer who participated through indirect means (e.g., by seeing who walks into the laboratory, by getting a list of who earned extra credit for participating in the study).
When recruiting from their own class is the only feasible way to do a study, researchers are expected to design the research in such a way that the potential for students to feel pressure is minimized. For example, if the research project concerns a teaching method that will be implemented in the course, then the only possible subject pool comes from the students enrolled in that course. If a research project has a reasonable chance of yielding benefits, and the only feasible way to complete the study is to recruit in the researcher’s course, the reach may be permissible if the researcher is able to sufficiently reduce the potential for students to feel pressure to participate.
References/Resources:
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 4.5 Researchers Recruiting from Their Own Courses.
Training-Related Topics
Who is required to complete the IRB human participants training?
University policy requires training for all faculty, faculty mentors, researchers, and students, including researchers from other institutions who are engaged in human subjects research at the University. See FAQ: What qualifies as Human Subjects Research requiring IRB Review? for the definition of engagement in human subjects research at UT Austin.
All personnel, originally listed or later added to a study through an amendment/modification, must complete human subjects research training course required by UT Austin. In order to comply with the policy, researchers are required to complete the University’s training affiliated with Collaborative Institutional Training Initiative (CITI). Completion of this training must be accomplished every three years.
References/Resources:
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 3.15 Training Requirements.
- IRB Training webpage.
Where can I find a list of my research team’s human subjects research training?
On the study’s main page within UTRMS-IRB, there is a link above the review timeline called “Study Staff Training Report” (Figure 7). Here, all listed study team members will have all their human subjects research training displayed should it be affiliated within our system.
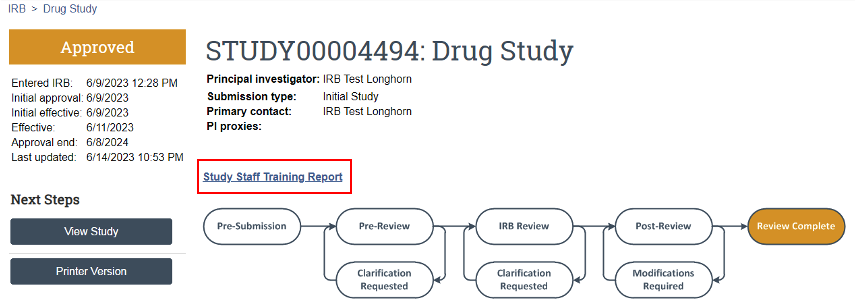
My training isn’t showing up or it is not up-to-date in the Study Staff Training Report; what do I do?
First, if you were added as the Primary Contact, make sure you were added as a study team member as well. Assigning Primary Contact does not automatically assign a researcher to the list of study team members.
Second, if you do not see your study staff training (for example, half of the boxes in the row are blank) or your updated training hasn’t showed up yet, please leave a Comment on the study with your name, UT EID, and CITI Member ID (not the CITI Record ID). The IRB Analyst assigned to your study will send in an affiliation request. The information should show up within 24 – 48 hours. If it does not, please contact the IRB at IRB@austin.utexas.edu.
After You Submit Your Proposal
What can I do if I have a question about an IRB determination?
Please contact your assigned IRB Analyst directly via comment within UTRMS, by email, or calling the number they provided, or contact the IRB Office at IRB@austin.utexas.edu.
Does my study require continuing review? How do I find this out?
Only studies with an approval end date/expiration date require a continuing review to be submitted to the IRB. Research determined to be minimal risk (receiving an exempt determination or expedited review) will not require annual continuing review unless specified by the IRB or additional federal regulations apply (e.g. FDA regulated).
To determine if a study has an approval end date or expiration date, visit the parent/main study page (Figure 8).
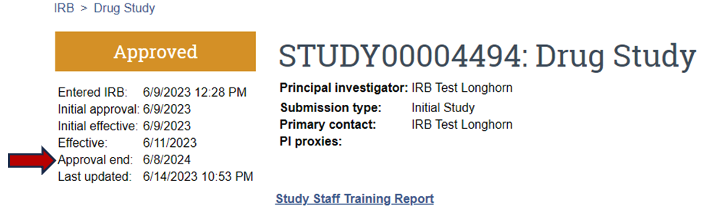
I received a "Notice of Study Lapse," what does this mean?
IRB approval for non-exempt research requiring a continuing review can be for no longer than a one-year period of time and there is no grace period beyond the expiration date of IRB approval. Extensions of approval beyond the expiration date cannot be granted. Failure to submit the required documents and receive IRB continuing approval for the protocol before the end of the approval period will result in a status of “Lapsed.” This will occur even if the PI has provided the required documents but IRB review and approval is not completed before the expiration date. If a protocol is placed in this status, the PI will be notified that they must cease all research activities (recruitment, advertisement, screening, enrollment, consent, interventions, interactions, and collection and analysis of private identifiable information) until the required documents are submitted, reviewed, and approved by the IRB.
References/Resources:
- UT Austin IRB’s HRP-UT1000 IRB Policies and Procedures Manual, Section 7.7 Failure to Comply with Continuing Review Requirements – Lapsed Protocols.
I received correspondence stating clarifications were requested, what does this mean?
This means that the assigned IRB Analyst has requested changes or clarification to your application or submitted documentation. The PI, PI Proxy and Primary Contact will receive an email notification advising them of the request. Details regarding the request can be found in the study workspace.
What If...?
I have complete access to everyone's records and want to use their information. Do I have to follow these guidelines?
No, appropriate authorization to obtain and use existing data for research purposes would still need to be obtained. For example, clinicians may have access to an individual’s HIPAA regulated protected health information (PHI) through the regular course of employment. When that PHI is accessed or used for research purposes, written HIPAA Authorization would need to be obtained (unless a waiver of HIPAA Authorization has been requested and granted) prior to its use for research.
What if I carried out my research without IRB approval?
University policy requires all research involving human subjects to be reviewed by ORSC or the IRB. If the study meets the definition of research involving human subjects, you must submit to the IRB. If you have conducted research involving human participants without IRB approval/determination, please contact the IRB office (IRB@austin.utexas.edu) to work with IRB support staff regarding appropriate reporting and determinations.
Conducting human subjects research without IRB approval/determination is considered noncompliance and requires appropriate corrective action and reporting.
Additional Information and What is Considered Human Subjects Research
Where can I read more on the regulations governing human subject research?
You can find a lot of information governing human subject research at the following sites:
- The Code of Federal Regulations:
- The U.S. Department of Health and Human Services, Office for Human Research Protections.
- The U.S. Department of Health and Human Services, National Institutes of Health.
- The U.S. Food and Drug Administration, Guidance Documents.
What happens when a non-research activity becomes research?
Research using data collected for non-research purposes requires IRB review to determine if the proposed research use meets the definitions of research involving human subjects (See FAQ: What qualifies as Human Subjects Research requiring IRB Review?). In this instance, HRP-UT903 – Template IRB Proposal Secondary Use Submission form would need to be used.
Is there anything else I should know?
All communication concerning general IRB questions should be submitted directly to the IRB Office at IRB@austin.utexas.edu. If your research activity has already been submitted, please reach out to your assigned IRB Analyst directly via comment within UTRMS, or by email.